Suture crânienne sur le front d'un enfant. Tout ce que vous devez savoir sur la fontanelle. Que menace-t-il ?
Le rôle décisif dans la formation et le développement ultérieur du crâne appartient au cerveau, aux dents, aux muscles masticateurs et aux organes sensoriels. Au cours du processus de croissance, la tête subit des changements importants. Au cours du développement, ils apparaissent âge, sexe et caractéristiques individuelles du crâne. Examinons quelques-uns d'entre eux.
Nouveau-nés
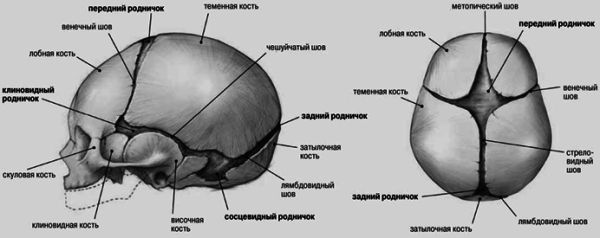
Le crâne du bébé a une structure spécifique. Les espaces entre les éléments osseux sont remplis de tissu conjonctif. Les nouveau-nés sont complètement absents sutures du crâne. Anatomie Cette partie du corps présente un intérêt particulier. A la jonction de plusieurs os se trouvent 6 fontanelles. Ils sont recouverts de plaques de tissu conjonctif. Il existe deux fontanelles non appariées (postérieure et antérieure) et deux appariées (mastoïdienne, sphénoïde). Le plus grand est considéré comme le frontal. Il a une forme de diamant. Il est situé à la convergence des os frontaux gauche et droit et des deux os pariétaux. Grâce aux fontanelles, il est très élastique. Lorsque la tête fœtale passe dans le canal génital, les bords du toit se chevauchent de manière carrelée. Pour cette raison, cela diminue. En règle générale, à l'âge de deux ans, ils ont formé sutures du crâne. Anatomieétudié auparavant de manière assez originale. Les médecins médiévaux appliquaient du fer chaud sur la zone de la fontanelle pour traiter les maladies des yeux et du cerveau. Après la formation d'une cicatrice, les médecins ont provoqué une suppuration avec divers irritants. Ils pensaient donc ouvrir la voie à l’accumulation de substances nocives. Dans la configuration des points de suture, les médecins ont essayé de discerner des symboles et des lettres. Les médecins pensaient qu'ils contenaient des informations sur le sort du patient.

Caractéristiques de la structure du crâne
Cette partie du corps d'un nouveau-né se distingue par la petite taille des os du visage. Une autre particularité concerne les fontanelles mentionnées ci-dessus. Dans le crâne d'un nouveau-né, il y a des traces des 3 étapes inachevées de l'ossification. Les fontanelles sont des vestiges de la période membraneuse. Leur présence est d'une importance pratique. Ils permettent aux os du toit de bouger. La fontanelle antérieure est située sur la ligne médiane à la jonction de 4 sutures : 2 moitiés coronaire, frontale et sagittale. Il devient envahi par la végétation au cours de la deuxième année de vie. La fontanelle postérieure a une forme triangulaire. Il est situé entre les deux en avant et les écailles de l'os occipital en arrière. Il repousse au cours du deuxième mois. Les fontanelles latérales se distinguent entre sphénoïde et mastoïde. Le premier est situé à la convergence des écailles pariétales, frontales, temporales et de la grande aile des os sphénoïdes. Pousse au cours du deuxième ou du troisième mois. La fontanelle mastoïde est située entre l'os pariétal, la base de la pyramide temporale et la squame occipitale.

Stade cartilagineux
À ce stade, les caractéristiques suivantes du crâne liées à l'âge sont notées. Des couches cartilagineuses se trouvent entre les éléments individuels non fusionnés des os de la base. Les sinus aériens ne sont pas encore développés. En raison de la faiblesse musculaire, diverses crêtes, tubercules et lignes musculaires sont mal définies. Pour la même raison, également associée au manque de fonction de mastication, les mâchoires sont sous-développées. Rarement. La mâchoire inférieure est constituée de deux moitiés non fusionnées. De ce fait, le visage s’étend peu vers l’avant par rapport au crâne. C'est seulement 1/8. De plus, chez un adulte, le rapport visage/crâne est de 1/4.
Déplacement osseux
Les crânes après la naissance se manifestent par une expansion active des cavités - nasale, cérébrale, buccale et nasopharynx. Cela entraîne un déplacement des os qui les entourent en direction des vecteurs de croissance. Le mouvement s'accompagne d'une augmentation de la longueur et de l'épaisseur. Avec une croissance marginale et superficielle, la courbure des os commence à changer.

Période postnatale
À ce stade, ils se manifestent par une croissance inégale des régions du visage et du cerveau. Les dimensions linéaires de ces dernières augmentent de 0,5 et celles des premières de 3 fois. Le volume de la région cérébrale double au cours des six premiers mois et triple au cours de la deuxième année. À partir de 7 ans, la croissance ralentit et s'accélère à nouveau lors de la puberté. Entre 16 et 18 ans, le développement de la voûte plantaire s'arrête. La base s'allonge jusqu'à 18-20 ans et se termine lorsque la synchondrose sphéno-occipitale se ferme. La croissance de la région faciale est plus longue et plus uniforme. Les os autour de la bouche se développent le plus activement. Caractéristiques d'âge du crâne au cours de la croissance, ils se manifestent par la fusion de parties d'os séparées chez le nouveau-né, une différenciation de structure et une pneumatisation. Le relief des surfaces internes et externes devient plus défini. Dès le plus jeune âge, des bords lisses se forment sur les coutures ; à l'âge de 20 ans, des articulations dentelées se forment.

Étapes finales
Vers l’âge de quarante ans, l’oblitération des sutures commence. Il couvre la totalité ou la plupart des connexions. À l'âge avancé et sénile, on observe une ostéoporose des os crâniens. L'amincissement des plaques de la substance compacte commence. Dans certains cas, on observe un épaississement des os. L'atrophie des mâchoires devient plus prononcée dans la région du visage en raison de la perte des dents. Cela provoque une augmentation de l’angle de la mâchoire inférieure. En conséquence, le menton avance.
Caractéristiques de genre
Il existe plusieurs critères selon lesquels un crâne masculin diffère de celui d'une femme. Ces signes incluent le degré de gravité de la rugosité et des tubercules dans les zones d'attache musculaire, le développement de la protubérance externe occipitale, la saillie de la mâchoire supérieure, etc. Le crâne masculin est plus développé que celui de la femme. Ses contours sont plus anguleux en raison de la sévérité des aspérités et des tubérosités au niveau des zones d'attache des muscles masticateurs, temporaux, occipitaux et cervicaux. Les tubercules frontaux et pariétaux sont plus développés chez la femme, tandis que chez l'homme, la glabelle et les arcades sourcilières sont plus développées. ces derniers ont une mâchoire inférieure plus lourde et plus grande. Au niveau du bord inférieur et des coins de la partie interne du menton, la tubérosité est clairement exprimée. Ceci est dû à l’attachement des muscles digastriques, masticatoires et ptérygoïdiens. Selon le sexe, la forme du crâne humain diffère également. Les hommes ont un front incliné qui se transforme en une couronne arrondie. Une élévation est souvent observée en direction de la suture sagittale. Le front des femmes est plus vertical. Elle se transforme en couronne plate. Les hommes ont des orbites inférieures. En règle générale, ils ont une forme rectangulaire. Leur bord supérieur est épaissi. Chez la femme, les orbites sont situées plus haut. Ils sont de forme presque ovale ou ronde avec des bords supérieurs plus nets et plus fins. Sur le crâne féminin, le processus alvéolaire fait souvent saillie vers l'avant. L'angle nasofrontal chez l'homme est clairement exprimé dans la plupart des cas. Sur le crâne féminin, l’os frontal passe plus facilement aux os nasaux.

En plus
La forme du crâne d’une personne n’affecte pas ses capacités mentales. Sur la base des résultats de nombreuses études menées par des anthropologues, nous pouvons conclure qu'il n'y a aucune raison de croire que la taille de la région cérébrale prédomine dans une race. Les Bushmen, les Pygmées et certaines autres tribus ont des têtes légèrement plus petites que les autres peuples. Cela est dû à leur petite taille. Souvent, une diminution de la taille de la tête peut être le résultat d'une mauvaise alimentation au fil des siècles et de l'influence d'autres facteurs défavorables.
La craniosténose est caractérisée par la fusion prématurée d'une ou plusieurs sutures crâniennes, entraînant souvent une forme anormale de la tête. Cela peut être le résultat d’une ossification anormale primaire (craniosténostose primaire) ou, plus communément, d’un trouble de la croissance du cerveau (craniosténostose secondaire).
La maladie survient souvent in utero ou à un âge très précoce. Elle ne peut être traitée que chirurgicalement, même si une issue positive n’est pas possible dans tous les cas.
Classification de la craniosténose et causes de son développement
L'ossification normale de la voûte crânienne commence dans la région centrale de chaque os du crâne et s'étend vers l'extérieur jusqu'aux sutures crâniennes. Qu'est-ce qui indique la normalité ?
- Lorsque la suture coronale sépare les deux os frontaux des os pariétaux.
- La suture métopique sépare les os frontaux.
- La suture sagittale sépare les deux os pariétaux.
- La suture lambdoïde sépare l'os occipital des deux os pariétaux.
Le principal facteur qui inhibe la fusion intempestive des os du crâne est la croissance continue du cerveau. Il convient de souligner que la croissance normale de chaque os crânien se produit perpendiculairement à chaque suture.
- La craniosténose simple est le terme utilisé dans les situations où une seule suture fusionne prématurément.
- Le terme craniosténose complexe ou jonctionnelle est utilisé pour décrire la fusion prématurée de plusieurs sutures.
- Lorsque les enfants présentant des symptômes de craniosténose souffrent également d’autres malformations corporelles, on parle de craniosténose syndromique.
Craniosynostose primaire
Si une ou plusieurs sutures subissent une fusion prématurée, la croissance du crâne peut être limitée par les sutures perpendiculaires. Si plusieurs sutures sont fusionnées alors que la taille du cerveau continue de changer, la pression intracrânienne peut augmenter. Et cela se termine souvent par un certain nombre de symptômes complexes, voire la mort.
Types de craniosténose primaire (fusion prématurée)
- La scaphocéphalie est une suture sagittale.
- La plagiocéphalie antérieure est la première suture coronale.
- La brachycéphalie est une suture coronale bilatérale.
- La plagiocéphalie postérieure est la fermeture précoce d'une suture lambdoïde.
- La trigonocéphalie est une fusion prématurée de la suture métopique.
Craniosynostose secondaire
Plus souvent que le type primaire, ce type de pathologie peut conduire à une fusion précoce des sutures en raison d'un échec primaire de la croissance cérébrale. Étant donné que la croissance du cerveau contrôle la distance entre les plaques osseuses, un trouble de sa croissance est la principale raison de la fusion prématurée de toutes les sutures.
Dans ce type de pathologie, la pression intracrânienne est généralement normale et une intervention chirurgicale est rarement nécessaire. Généralement, le manque de croissance cérébrale conduit à une microcéphalie. La fermeture prématurée de la suture, qui ne constitue pas une menace pour la croissance du cerveau, ne nécessite pas non plus d'intervention chirurgicale.
Les restrictions spatiales intra-utérines peuvent jouer un rôle dans la fusion prématurée des sutures du crâne fœtal. Cela a été démontré dans des cas de craniosténose coronale. D'autres causes secondaires incluent des troubles systémiques affectant le métabolisme osseux tels que le rachitisme et l'hypercalcémie.
Causes et conséquences de la craniosténose précoce
Plusieurs théories ont été proposées pour l'étiologie de la craniosténose primaire. Mais l'option la plus répandue est celle avec l'étiologie d'un défaut primaire des couches mésenchymateuses des os du crâne.
La craniosténose secondaire se développe généralement en même temps que des troubles systémiques
- Il s'agit de troubles endocriniens (hyperthyroïdie, hypophosphatémie, carence en vitamine D, ostéodystrophie rénale, hypercalcémie et rachitisme).
- Maladies hématologiques provoquant une hyperplasie de la moelle osseuse, par exemple la drépanocytose, la thalassémie.
- Faibles taux de croissance cérébrale, y compris la microcéphalie et ses causes sous-jacentes, telles que l'hydrocéphalie.

Les causes de la craniosténose syndromique sont des mutations génétiques responsables des récepteurs du facteur de croissance des fibroblastes des deuxième et troisième classes.
Autres facteurs importants à prendre en compte lors de l'étude de l'étiologie de la maladie
- Différencier la plagiocéphalie, qui est souvent le résultat d’une fusion positionnelle (qui ne nécessite pas de chirurgie et est assez courante) de la fusion par suture lambdoïde, est un aspect extrêmement important.
- La présence d’adhérences multiples est évocatrice d’un syndrome cranio-facial, qui nécessite souvent une évaluation diagnostique en génétique pédiatrique.
Symptômes de la craniosténose et méthodes de diagnostic
La craniosténose est caractérisée dans tous les cas par une forme irrégulière du crâne, qui chez un enfant est déterminée par le type de craniosténose.
Caractéristiques principales
- Une crête osseuse rigide, facilement palpable le long de la suture pathologique.
- La zone molle (fontanelle) disparaît, la tête de l'enfant change de forme et la sensibilité de ces zones est généralement altérée.
- La tête du bébé ne grandit pas proportionnellement au reste du corps.
- Augmentation de la pression intracrânienne.
Dans certains cas, la craniosténose peut n’être perceptible que plusieurs mois après la naissance.
L'augmentation de la pression intracrânienne est une caractéristique commune à tous les types de craniosténose, à l'exception de certaines pathologies secondaires. Lorsqu’une seule suture fusionne prématurément, une augmentation de la pression intracrânienne survient chez moins de 15 % des enfants. Cependant, dans les craniosténoses syndromiques, où plusieurs sutures sont impliquées, une augmentation de la pression peut être observée dans 60 % des cas.
Si un enfant souffre d'une forme légère de craniosténose, la maladie peut ne pas être remarquée jusqu'à ce que les patients commencent à éprouver des problèmes dus à une augmentation de la pression intracrânienne. Cela se produit généralement entre quatre et huit ans.
Symptômes d'augmentation de la pression intracrânienne
- Ils commencent par des maux de tête persistants, généralement pires le matin et le soir.
- Problèmes de vision - vision double, vision floue ou vision des couleurs altérée.
- Déclin inexpliqué des capacités mentales de l'enfant.
Si votre enfant se plaint de l'un des symptômes ci-dessus, vous devez contacter votre pédiatre dès que possible. Dans la plupart des cas, ces symptômes ne sont pas causés par une augmentation de la pression intracrânienne, mais ils doivent absolument être étudiés.
S'ils ne sont pas traités, d'autres symptômes d'augmentation de la pression intracrânienne peuvent inclure :
- vomissement;
- irritabilité;
- léthargie et manque de réponse ;
- yeux enflés ou difficulté à voir un objet en mouvement.
- déficience auditive;
- respiration difficile.
En examinant de plus près le crâne, il apparaît clairement que sa forme ne confirme pas toujours le diagnostic de craniosténose. Dans de tels cas, un certain nombre de méthodes d'examen visuel sont utilisées, par exemple une radiographie du crâne.
Les radiographies sont réalisées dans plusieurs projections : antérieure, postérieure, latérale et supérieure. Les sutures fusionnées prématurément sont facilement identifiées par l’absence de lignes connectées et la présence de crêtes osseuses le long de la ligne de suture. Les sutures elles-mêmes ne sont pas visibles ou leur emplacement montre des signes de sclérose.
Une tomodensitométrie crânienne 3D n’est généralement pas nécessaire pour la plupart des nourrissons. Cette technique est parfois réalisée lorsque la chirurgie est envisagée comme prochaine étape du traitement ou lorsque les résultats radiographiques sont équivoques.
Méthodes de correction de la pathologie, complications possibles et conséquences

Au cours des 30 dernières années, la médecine moderne a développé une meilleure compréhension de la physiopathologie et du traitement de la craniosténose. Actuellement, la chirurgie reste généralement le principal type de traitement pour la correction des déformations crâniennes chez les enfants présentant des fusions de 1 à 2 sutures, entraînant une tête malformée. Pour les enfants atteints de microcéphalie, souvent associée à une craniosténose légère, la chirurgie n'est généralement pas nécessaire.
Lors de l'élaboration d'un schéma thérapeutique, les spécialistes doivent prendre en compte un certain nombre de points.
- Les patients atteints de microcéphalie devraient avoir la cause de cette maladie a été étudiée.
- Lors de votre premier contact le tour de tête est mesuré dans le sens longitudinal et plus loin les changements sont surveillés. Le médecin doit vérifier la croissance cérébrale normale chez les patients atteints de craniosténose primaire.
- A effectuer régulièrement surveillance des signes et symptômes d'augmentation de la pression intracrânienne.
- S'il y a une suspicion d'augmentation de la pression intracrânienne, alors c'est très approprié consultation de neurochirurgie.
- Pour préserver la fonction visuelle chez les patients présentant une pression intracrânienne accrue, il est nécessaire d'effectuer consultations ophtalmologiques complémentaires.
La chirurgie est généralement prévue en cas d’augmentation de la pression intracrânienne ou pour corriger une déformation crânienne. L’opération est généralement réalisée au cours de la première année de vie.
Conditions pour la chirurgie
- Si la forme de la tête ne s'améliore pas à l'âge de deux mois, il est peu probable que l'anomalie change avec l'âge. Une intervention précoce est indiquée si les enfants peuvent être candidats à une chirurgie mini-invasive. Il convient de noter que la déformation est plus visible au cours de la période thoracique et qu’elle peut devenir moins évidente avec l’âge.
- À mesure que l’enfant grandit et développe davantage de poils, les signes visibles de l’anomalie peuvent diminuer.
- Les indications de correction chirurgicale de la craniosténose dépendent de l'âge, de l'état général de l'enfant et du nombre de sutures fusionnées prématurément.
- Le traitement chirurgical de la déformation crânienne ou cranio-faciale est pratiqué chez les enfants âgés de 3 à 6 mois, bien que les approches varient selon les chirurgiens.

La chirurgie chez les nourrissons peut entraîner des pertes de volume sanguin relativement importantes. En conséquence, des techniques chirurgicales mini-invasives doivent être envisagées. Une option prometteuse est l’utilisation d’acide tranexamique peropératoire. Les patients présentant des indications pour une correction chirurgicale de la craniosténose ont été prétraités avec de l'érythropoïétine et de l'acide tranexamique, ce qui leur a permis de maintenir des pertes sanguines plus faibles.
Autres caractéristiques de la chirurgie
- La chirurgie chez les nourrissons de plus de 8 mois peut être associée à un ralentissement de la croissance du crâne.
- Les nourrissons diagnostiqués avec une craniosténose syndromique devraient subir une intervention chirurgicale dès que possible.
- Les résultats de l’opération sont meilleurs si elle est réalisée sur des nourrissons de moins de 6 mois.
Craniosténose - (du grec kranion - crâne et sténose - rétrécissement) fermeture prématurée des sutures crâniennes ou leur absence congénitale.
Le crâne de bébé est constitué de 6 os du crâne : l'os frontal, l'os occipital, deux os pariétaux, deux os temporaux. Normalement, tous ces os du crâne ne sont pas fusionnés, les fontanelles antérieure et postérieure sont ouvertes. Les os énumérés ci-dessus sont maintenus ensemble par des tissus solides et élastiques appelés sutures. Sans la flexibilité de ces sutures, le cerveau d’un enfant ne peut pas se développer correctement. Le cerveau grandit et la paupière du crâne de l'enfant devrait également se dilater. Les sutures répondent à la croissance du cerveau en « étirant » et en « produisant » de nouveaux os, permettant ainsi au crâne de croître avec le cerveau. La croissance normale du crâne se produit perpendiculairement à chaque suture.
La fontanelle postérieure se ferme à la fin du 2ème mois, la fontanelle antérieure - au cours de la 2ème année de vie. Vers la fin du 6ème mois de vie, les os de la voûte crânienne sont reliés entre eux par une membrane fibreuse dense. À la fin de la première année de vie, la taille de la tête de l’enfant atteint 90 % et à 6 ans, elle atteint 95 % de la taille de la tête de l’adulte. La fermeture des sutures en reliant les bords irréguliers des os commence vers la fin de la 1ère année de vie et se termine complètement vers l'âge de 12-14 ans.
La prolifération prématurée et inégale des fontanelles et des sutures crâniennes chez les enfants - craniosténose - interfère avec le développement normal du cerveau et conduit à la création de conditions propices à des troubles de la dynamique de l'alcool. Les troubles liquorodynamiques sont généralement appelés états pathologiques dans lesquels la sécrétion, la résorption et la circulation du liquide céphalo-rachidien sont altérées. L'incidence de la craniosténose est de 1 nouveau-né sur 1 000. Avec la craniosténose, la pression intracrânienne est généralement augmentée, c'est pourquoi des maux de tête hypertensifs sont caractéristiques et des disques optiques congestifs peuvent se développer, suivis d'une atrophie secondaire, d'une déficience visuelle et d'un retard mental.
Causes
Il existe des craniosténoses primaires (idiopathiques) et secondaires. Le développement d'une craniosténose secondaire peut être dû à diverses raisons. Ceux-ci peuvent inclure le rachitisme par carence en vitamine D, l'hypophosphatémie et le surdosage d'hormones thyroïdiennes en cas de traitement de l'oligophrénie hypothyroïdienne congénitale (crétinisme).
Ce qui se passe?
La cicatrisation des sutures du crâne est non seulement prématurée, mais aussi inégale et conduit généralement à une déformation du crâne. Dans le processus de surveillance de l'évolution de la forme du crâne cérébral, ce que l'on appelle l'indice crânien (IC) est pris en compte - le rapport entre la taille transversale du crâne et sa taille longitudinale, multiplié par 100. Avec une normale Rapport (moyen) des dimensions transversales et longitudinales de la tête (avec mésocéphalie), l'indice crânien chez les hommes est de 76 à 80,9, pour les femmes de 77 à 81,9.
En cas de prolifération prématurée de la suture sagittale (synostose sagittale), dolichocéphalie, dans lequel le crâne augmente dans la direction antéropostérieure et diminue en taille transversale. Dans de tels cas, la tête s'avère étroite et allongée. Le CHI est inférieur à 75.
 Une variante de dolichocéphalie causée par une fusion prématurée de la suture sagittale, dans laquelle il existe une restriction de la croissance du crâne dans le sens transversal et une croissance excessive en longueur, peut être scaphocéphalie(du grec skaphe - bateau), cymbocéphalie (tête scaphoïde, tête de quille), dans laquelle se forme une tête longue et étroite avec un front et un arrière de la tête saillants, rappelant un bateau renversé avec sa quille. Un crâne allongé dans le sens longitudinal avec une dépression dans la région pariétale est appelé en forme de selle.
Une variante de dolichocéphalie causée par une fusion prématurée de la suture sagittale, dans laquelle il existe une restriction de la croissance du crâne dans le sens transversal et une croissance excessive en longueur, peut être scaphocéphalie(du grec skaphe - bateau), cymbocéphalie (tête scaphoïde, tête de quille), dans laquelle se forme une tête longue et étroite avec un front et un arrière de la tête saillants, rappelant un bateau renversé avec sa quille. Un crâne allongé dans le sens longitudinal avec une dépression dans la région pariétale est appelé en forme de selle.
Une variante de déformation du crâne, dans laquelle le crâne a une taille transversale accrue en raison d'une fusion prématurée des sutures coronales (coronales) (synostose coronale ou coronale), est brachycéphalie(du grec brachis - court et képhale - tête), la tête est large et raccourcie, l'index crânien est supérieur à 81. Avec la brachycéphalie, due à une synostose coronarienne bilatérale, le visage est aplati, une exophtalmie se manifeste souvent - déplacement antérieur d'un ou les deux globes oculaires.
Avec fusion prématurée de la suture coronaire d'un côté, plagiocéphalie, ou tête croisée (du grec plagios - oblique et kephale - tête). Dans de tels cas, le crâne est asymétrique, l'os frontal du côté de la synostose est aplati et une exophtalmie et une hypertrophie des fosses crâniennes moyennes et postérieures sont possibles du même côté.
 En cas de fusion prématurée combinée des sutures crâniennes coronales et sagittales, la croissance du crâne se fait principalement vers la fontanelle antérieure et la base, ce qui entraîne une augmentation de la hauteur de la tête tout en limitant sa croissance dans les directions longitudinale et transversale. En conséquence, un crâne haut et conique se forme, quelque peu aplati dans la direction antéropostérieure (acrocranie) ; on l'appelle souvent un crâne en forme de tour. Une variante du crâne de la tour est l'oxycéphalie, ou tête pointue (du grec oxys - pointu, képhale - tête), dans laquelle la fusion précoce des sutures crâniennes conduit à la formation d'un crâne haut et effilé vers le haut avec un front incliné vers l'arrière.
En cas de fusion prématurée combinée des sutures crâniennes coronales et sagittales, la croissance du crâne se fait principalement vers la fontanelle antérieure et la base, ce qui entraîne une augmentation de la hauteur de la tête tout en limitant sa croissance dans les directions longitudinale et transversale. En conséquence, un crâne haut et conique se forme, quelque peu aplati dans la direction antéropostérieure (acrocranie) ; on l'appelle souvent un crâne en forme de tour. Une variante du crâne de la tour est l'oxycéphalie, ou tête pointue (du grec oxys - pointu, képhale - tête), dans laquelle la fusion précoce des sutures crâniennes conduit à la formation d'un crâne haut et effilé vers le haut avec un front incliné vers l'arrière.
Une variante de déformation du crâne, caractérisée par des os frontaux étroits et des os occipitaux larges, se forme en raison de la fusion prématurée de la suture frontale. Dans ce cas, les os frontaux se rapprochent selon un angle (normalement, la suture frontale ne guérit qu'à la fin de la 2e année de vie) et une « crête » se forme à l'emplacement de la suture frontale. Si, dans de tels cas, les parties postérieures du crâne s'agrandissent de manière compensatoire et que sa base s'approfondit, une trigonocranie, ou un crâne triangulaire, apparaît (du grec trigonon - triangle, képhale - tête).
La synostose isolée de la suture lambdoïde est extrêmement rare et s'accompagne d'un aplatissement de l'occiput et d'une expansion compensatoire de la partie antérieure du crâne avec un élargissement de la fontanelle antérieure. Elle est souvent associée à une fermeture prématurée de la suture sagittale.
À craniosténose secondaireà un stade précoce de son développement, un traitement conservateur de la maladie sous-jacente peut être efficace. En cas de craniosténose primaire, ainsi qu'en cas de craniosténose secondaire en cas d'hypertension intracrânienne importante déjà développée, une chirurgie décompressive est indiquée : formation de passages de craniectomie jusqu'à 1 cm de large le long de la ligne d'ossifications de suture. Un traitement chirurgical opportun de la craniosténose peut assurer un développement cérébral normal.
Traitement
La période la plus active de croissance cérébrale est considérée comme allant jusqu’à l’âge de deux ans. Ainsi, d’un point de vue fonctionnel, la craniosténose peut être prévenue par un traitement chirurgical précoce. L'âge optimal pour la chirurgie de la craniosténose peut être considéré comme la période de 3 à 9 mois. Les avantages du traitement à cet âge comprennent : la facilité de manipulation des os fins et mous du crâne ; faciliter le remodelage final de la forme du crâne par le cerveau en croissance rapide ; une guérison plus complète et plus rapide des défauts osseux résiduels.
Si le traitement est effectué après l’âge de cinq ans, il est peu probable qu’il entraîne une amélioration significative des fonctions cérébrales. Dans une plus large mesure, l'opération visera à éliminer la déformation de la tête. La principale caractéristique du traitement chirurgical moderne n'est pas seulement l'augmentation du volume du crâne, mais également la correction de sa forme et de la déformation faciale associée au cours d'une opération.
À quoi les parents doivent prêter attention
- Forme inhabituelle de la tête d'un enfant
- Fermeture anticipée d'une grande fontanelle (jusqu'à un an)
- Le taux de croissance du tour de tête de l’enfant ne correspond pas à la norme d’âge (voir le tour de tête des garçons et le tour de tête des filles)
- Mauvais sommeil, agitation de l'enfant, détérioration de l'enfant lorsque le temps change, régurgitation, retard du développement psychomoteur (voir développement psychomoteur de l'enfant)
Si les symptômes ci-dessus sont détectés chez un enfant, vous devez contacter un spécialiste :
- Neuropathologiste – évalue la présence de symptômes neurologiques et d'un retard de développement de l'enfant
- Ophtalmologiste - évalue les signes d'hypertension intracrânienne sur la base des résultats de l'examen du fond d'œil (dans les cas avancés - diminution de l'acuité visuelle)
- Pédiatre – évalue la présence d'autres anomalies dans le développement des organes et des systèmes, pathologie somatique
- Génétique – révèle la présence de la nature génétique de la maladie, la probabilité d'anomalies dans d'autres organes et systèmes et le pronostic de la récidive d'une pathologie similaire chez le prochain enfant
Attention, il vaut mieux jouer la sécurité et orienter un enfant présentant une déformation du crâne vers un spécialiste plutôt que de passer à côté de la pathologie.
Avez-vous aimé l'article? Partagez le lien
Le crâne d’un nouveau-né est constitué de six os distincts reliés par de fines couches élastiques de tissu fibreux. Dans la pratique médicale, on les appelle sutures crâniennes ou fontanelles.
De telles zones sont nécessaires pour que les os se chevauchent partiellement pendant le processus d'accouchement. Cette nuance permet exclure une lésion cérébrale nouveau née
Les fontanelles disparaissent chez l'enfant au cours de la première année de vie. La fusion prématurée des tissus mous est appelée craniosténose (craniosténose) et est une pathologie grave.
Concept et caractéristiques
Craniosténose chez un enfant - photo :
La craniosténose est une pathologie dans laquelle une fermeture précoce ou une absence totale de sutures crâniennes se produit chez les enfants. Cette condition entraîne une hypertension intracrânienne, une déformation importante du crâne et une limitation de son volume.
La pathologie peut être primaire ou secondaire. Dans le premier cas, la maladie se développe dans le contexte d'une formation inappropriée du squelette osseux et d'une ossification du crâne; dans le second, la perturbation du processus de croissance cérébrale joue un rôle important.
Caractéristiques de la maladie:
- la pathologie commence à progresser au stade du développement intra-utérin du fœtus;
- dans de rares cas, la maladie se manifeste chez les nouveau-nés au cours des premiers mois de la vie ;
- si la pathologie n'a pas été détectée à la naissance ou dans les premiers mois de la vie, le risque de son développement à un âge plus avancé est exclu.
Causes
La base du processus pathologique est une violation de la formation du squelette osseux fœtal.
Raisons du développement de cette condition dans la pratique médicale pas complètement étudié, mais les experts identifient plusieurs facteurs qui augmentent le risque de craniosténose chez les nouveau-nés.

Dans la plupart des cas, la pathologie est associée à d'autres défauts du développement de l'enfant.
Causes de pathologie sont les facteurs suivants :
- pathologies héréditaires (par exemple, syndrome de Crouzon) ;
- conséquences d'un déséquilibre hormonal ;
- pathologies endocriniennes graves ;
- la position du fœtus dans laquelle son crâne est comprimé par l'utérus ;
- synostose prématurée d'une ou plusieurs sutures crâniennes ;
- abus de mauvaises habitudes pendant la grossesse;
- utilisation incontrôlée de médicaments puissants pendant la gestation ;
- position anormale du fœtus pendant la grossesse ;
- croissance cérébrale lente;
- traumatisme périnatal;
- maladies hématologiques;
- anomalies génétiques;
- exposition à des infections intra-utérines.
Quels sont les signes du syndrome de Down chez un nouveau-né ? découvrez-le dès maintenant.
Classification : types et formes
Dans la pratique médicale, la classification de la craniosténose s'effectue dans plusieurs directions. La pathologie peut être syndromiques et non syndromiques. Dans le premier cas, la maladie s'accompagne d'autres défauts, dans le second, elle se développe indépendamment.

Selon le nombre de sutures fusionnées, la craniosténose est divisée en pansynostose(fusion de toutes les sutures du crâne), monosynostose(épissage d'une couture) et polysynostose(fusion de plusieurs coutures).
La monosynostose est divisée en types distincts :
- lambdoïde craniosynostose unilatérale ou bilatérale (fusion prématurée de la suture lambdoïde) ;
- isolémétopique craniosténose (prolifération de la suture métopique) ;
- coronaire craniosténose unilatérale ou bilatérale (fusion de la suture coronale) ;
- sagittal isolé Craniosynostose (fusion prématurée de la suture sagittale).
Symptômes et signes
Les symptômes de la craniosténose dépendent directement du stade de progression du processus pathologique et du nombre de sutures fermées. La période de formation fœtale pendant laquelle cette pathologie se manifeste joue un rôle important.
Si les sutures commencent à se refermer dès les premiers mois de la grossesse, les symptômes seront plus prononcés.
Les signes externes de craniosténose apparaissent immédiatement après la naissance de l'enfant et clairement visible visuellement.
Symptômes pathologie sont les facteurs suivants :

Diagnostique
Le diagnostic de craniosténose chez un enfant commence par un examen visuel. Le médecin palpe les fontanelles, détecte les écarts par rapport à la norme et recueille un historique médical complet. Les paramètres du crâne du bébé sont mesurés à l'aide ruban spécial.
Sur la base des données collectées, le spécialiste prescrit des tests, procédures et consultations complémentaires avec des médecins spécialisés. L'examen du nouveau-né doit être effectué de manière exhaustive et inclure identification d'éventuelles pathologies supplémentaires.
Diagnostique La craniosténose comprend les procédures suivantes :
- IRM et tomodensitométrie du cerveau ;
- angiographie;
- examen neurologique;
- examen physique;
- Échographie Doppler des vaisseaux du cou et de la tête ;
- neurosonographie;
- consultation avec un ophtalmologiste;
- ophtalmoscopie;
- Radiographie du cerveau.
Méthodes de traitement

Lors du traitement de la craniosténose dans la pratique médicale, deux méthodes principales sont utilisées - chirurgie et endoscopie.
Les moyens radicaux de corriger la maladie sont les plus efficaces.
Endoscopie n'est réalisée que jusqu'à l'âge de six mois de l'enfant et en présence de complications minimes du processus pathologique. La décision d'utiliser une méthode de traitement spécifique pour la craniosténose est prise par le médecin sur la base d'un examen complet du bébé.
Régime thérapeutique
Le traitement thérapeutique de la craniosténose comprend plusieurs étapes. Après avoir examiné l'enfant, le médecin détermine le degré de développement de la maladie et identifie la présence de complications du processus pathologique.
Il existe certaines contre-indications à l'intervention chirurgicale.
Par exemple, une période d'exacerbation d'une maladie virale, des maladies liées à la composition du sang ou le bébé a une forte fièvre. Après l'examen un type spécifique d'intervention chirurgicale est sélectionné.
Schéma thérapeutique pour la craniosténose :

Chirurgical
L'intervention chirurgicale est le moyen principal et le plus efficaceélimination de la craniosténose.
Le but de cette procédure est de donner au crâne la forme correcte en coupant les sutures fusionnées prématurément.
Dans certains cas, les spécialistes utilisent des dispositifs de distraction pour remodeler les os.
Caractéristiques de l'opération:
- Il est recommandé d’effectuer la procédure jusqu’à sept mois (la croissance active du cerveau se produit au cours de la première année de la vie d’un enfant).
- Une intervention chirurgicale précoce permet une régression accrue de toutes les conditions pathologiques existantes chez l'enfant.
- Une opération opportune augmente considérablement la probabilité d'un pronostic favorable pour le bébé.
Vous trouverez des informations sur le traitement de l'hydrocéphalie chez un enfant sur notre site Internet.
Prévisions
Craniosynostose chez les enfants - photos avant et après :

Le pronostic de la craniosténose dépend de la forme de la maladie présente. Éliminer presque complètement les conséquences la maladie ne peut être traitée qu'en temps opportun.
Dans certains cas, les complications entraînent la mort de l'enfant au cours de la première année de vie.
Les enfants sont d'abord infectés par les ARVI, puis, sur fond de rhume, ils développent une pneumonie. Mauvais pronostic observé uniquement dans la forme syndromique de craniosténose.
Nuances pronostic pour pathologie :
- Plus l’intervention chirurgicale est réalisée tôt, plus le risque de complications est faible.
- L'endoscopie s'accompagne de moins de pertes de sang pour l'enfant (la procédure a des restrictions d'âge).
- Si l'opération est réalisée après que l'enfant ait atteint l'âge de trois ans, le risque de complications sera maximum.
Mesures spécifiques pour prévenir la craniosténose chez les enfants n'existe pas.
Les actions préventives non spécifiques comprennent l'attitude attentive d'une femme à l'égard de sa propre santé et de la période de grossesse, des examens réguliers par un gynécologue, ainsi qu'une prévention rapide du risque de développer des infections intra-utérines.
Si un enfant reçoit un diagnostic de craniosténose, un traitement de la maladie est alors nécessaire. effectuer dans les plus brefs délais.
Vous pouvez en apprendre davantage sur le pronostic de la craniosténose chez les enfants grâce à la vidéo :
Nous vous demandons de ne pas vous soigner vous-même. Prenez rendez-vous avec un médecin !
La craniosténose est une pathologie de cicatrisation des sutures du crâne, qui provoque une déformation de la forme normale de la tête. La synostose métopique (trigonocéphalie) est le type de craniosynostose le plus rare, représentant environ 10 à 25 % de ces maladies.
La synostose métopique chez un enfant est généralement diagnostiquée à la maternité. Des formes bénignes peuvent être détectées lors d'un examen par un néonatologiste ou un pédiatre (à un âge plus avancé). La trigonocéphalie est traitée chirurgicalement.
La synostose métopique résulte d'une fusion prématurée du crâne dans la zone de la suture métopique. Cette structure anatomique relie les parties de l'os frontal. Il est considéré comme normal que la suture métopique se ferme dans un délai de 8 à 24 mois. La fermeture intempestive de la couture provoque une déformation du crâne, ainsi que de la partie supérieure du visage.
Il existe deux types de synostose métopique : la trigonocéphalie et la craniosténose asymptomatique. Le premier type de déformation présente les caractéristiques suivantes :
- La fermeture des sutures se produit généralement in utero ou dans la petite enfance (jusqu'à 2 mois) ;
- La déformation se forme selon le type d'hypotélorisme ou de trigonocéphalie ;
- Cliniquement, cela se manifeste par une diminution de la distance entre les orbites ;
- L'emplacement de la prolifération peut être déterminé par palpation sous la forme d'une petite bosse ;
- La croissance complète et le lissage prennent jusqu'à 2 mois.
La fermeture précoce de la suture métopique entraîne un développement insuffisant de l'os frontal. Visuellement, cela se caractérise par un visage ovale disproportionné, mais relativement petit.
Type 2 (asymptomatique)  Craniosynostose) se développe relativement plus tard. La cicatrisation de la suture mépotique est observée à l'âge de 3-4 mois. Une crête osseuse se forme à la jonction, détectable par palpation. La craniosynostose asymptomatique ne déforme pas l'os frontal et l'ovale du visage, le diagnostic visuel est donc difficile.
Craniosynostose) se développe relativement plus tard. La cicatrisation de la suture mépotique est observée à l'âge de 3-4 mois. Une crête osseuse se forme à la jonction, détectable par palpation. La craniosynostose asymptomatique ne déforme pas l'os frontal et l'ovale du visage, le diagnostic visuel est donc difficile.
Caractéristiques générales de la maladie
La synostose métopique chez un enfant est une pathologie relativement rare de ce groupe de maladies. Ces dernières années, on a observé une tendance à l'augmentation de l'incidence de la maladie dans la population en raison de l'influence accrue de facteurs tératogènes pendant la grossesse (radiations, médicaments, mauvaise alimentation). Le déclencheur du développement de la maladie peut être une blessure au crâne lors du passage de l'enfant dans le canal génital.
Le tableau clinique de la synostose métopique est caractérisé par :
- Syndrome d'hypertension ;
- Les troubles mentaux;
- Pathologie du nerf optique.
L'hypertension est causée par une augmentation de la pression à l'intérieur du crâne. En raison du rétrécissement pathologique de l'espace, les vaisseaux sanguins n'ont pas la possibilité de se développer comme d'habitude. Cela peut provoquer une perturbation de la circulation locale entraînant une ischémie ultérieure.

La croissance la plus active du cerveau principal et des neurones se produit sur une période pouvant aller jusqu'à deux ans. La fermeture prématurée de la suture métopique entraîne un manque d'espace pour la croissance de la matière grise, de sorte que la trigonocéphalie conduit à un handicap mental. Dès le plus jeune âge, la pathologie du développement de cette zone est réversible. Elle est corrigée par la chirurgie.
La réduction de la distance entre les orbites contribue à la compression du nerf optique. Les impulsions traversant ces faisceaux sont déformées, ce qui conduit à l'apparition de pathologies ophtalmiques.
Le diagnostic de synostose métopique peut être posé par un pédiatre lors d'un examen de routine de l'enfant. En plus de la forme caractéristique du visage, lors de la palpation de la zone de suture envahie, une crête d'origine inerte peut être détectée. Les symptômes neurologiques de la trigonocéphalie sont difficiles à déterminer, le diagnostic repose donc sur la vérification des réflexes lors de l'examen par un neurologue.
La tomodensitométrie est utilisée pour effectuer un diagnostic différentiel. La méthode permet de vérifier la présence de disproportions dans la région cranio-cérébrale, ainsi que de confirmer la présence de compression des parties frontales du cerveau dans le sens sagittal.
Traitement de la synostose métopique
La trigonocéphalie est une indication chirurgicale.

Un diagnostic rapide et un traitement immédiat réduisent au minimum le risque de complications. La période optimale pour une intervention chirurgicale est considérée comme comprise entre 3 et 9 mois. Les structures osseuses pendant cette période sont relativement labiles, de sorte que l'opération n'est pas compliquée par la nécessité d'une craniotomie.
La croissance rapide du cerveau durant cette période contribue à l'égalisation rapide des déformations sans conséquences pour le système nerveux. Les défauts indirects guérissent complètement, ne laissant presque aucune cicatrice, et la période de rééducation est relativement meilleure.
Une caractéristique de l'intervention chirurgicale pour la synostose métopique est une augmentation du volume du crâne ainsi qu'une modification de sa forme. Cela élimine la déformation de la région du visage. Les capacités de régénération du système nerveux dépendent de l’âge.
Plus l'état du patient est facile, moins il y aura de complications après l'opération. L'intervention chirurgicale après 5 ans n'est pas très efficace. Cette opération n'améliorera pas les fonctions cognitives.
Sans chirurgie, la trigonocéphalie progressera avec le temps. Une déformation profonde du crâne sans la chirurgie plastique nécessaire peut entraîner une incapacité mentale complète et une détérioration de la vision. Après une intervention chirurgicale pendant la rééducation, le recours à des procédures physiothérapeutiques est recommandé.
La synostose métopique est une forme sévère de déformation du crâne dans le sens sagittal. Cette pathologie peut entraîner un certain nombre de complications. Un diagnostic rapide de la maladie joue un rôle important.

Après avoir détecté des symptômes suspects, vous devez en informer votre pédiatre ou contacter vous-même un neurologue. Il ne faut pas oublier que plus l’intervention chirurgicale est pratiquée tôt, moins il y aura de conséquences à l’avenir.









